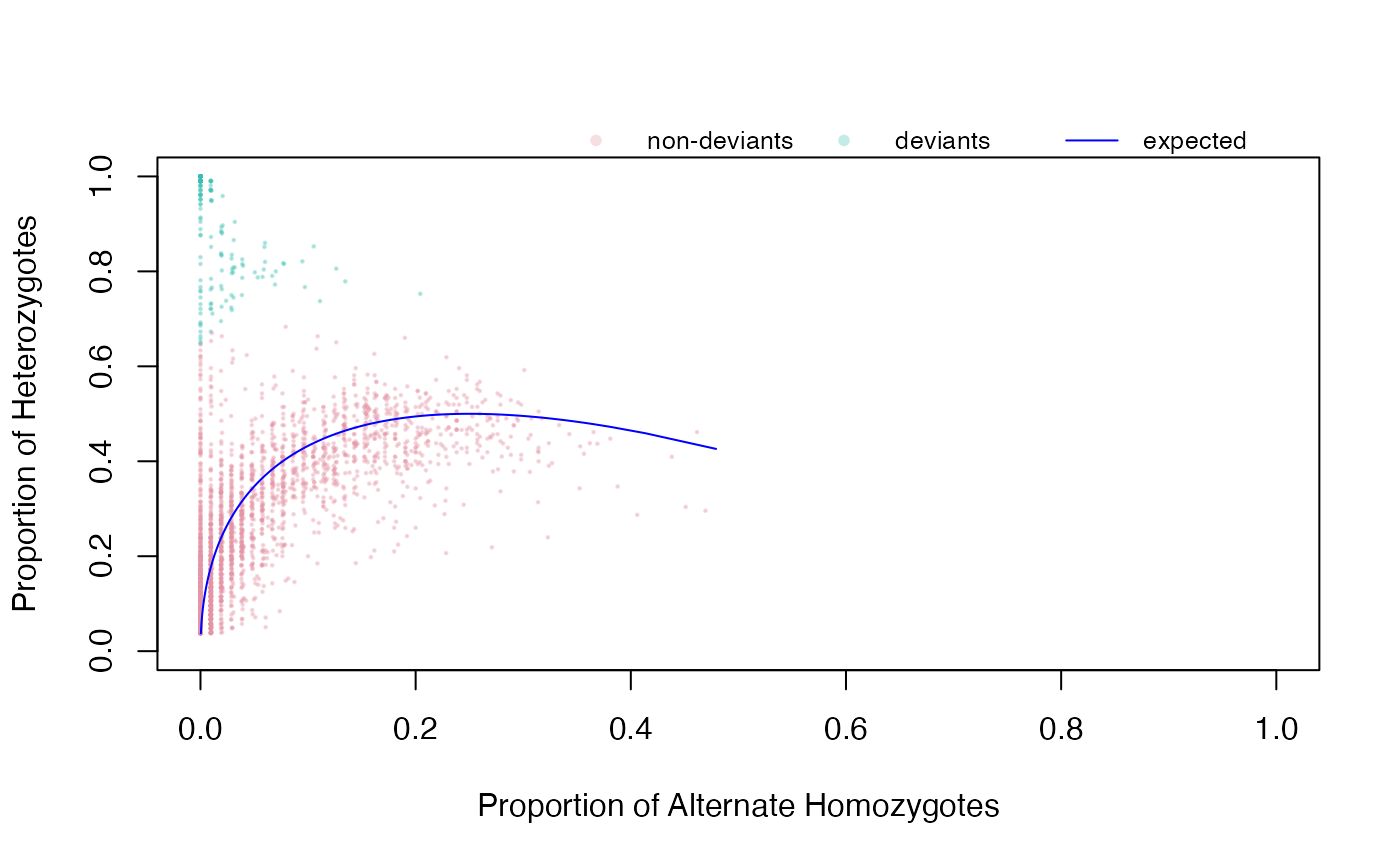
This function will recognize the SNPs with a proportion of heterozygotes significantly higher than expected under HWE and plot deviant snps based only on the excess of heterozygotes.
Usage
sig.hets(
a.info,
Fis,
method = c("chi.sq", "fisher"),
plot = TRUE,
verbose = TRUE,
...
)Arguments
- a.info
allele info table generated from filtered vcfs using the function
allele.infoor allele depth table generated fromhetTgen- Fis
numeric. Inbreeding coefficient calculated using
h.zygosity()function- method
character. Method for testing significance. Fisher exact test (
fisher) or Chi-square test (chi.sq)- plot
logical. Whether to plot the identified duplicated snps with the expected values
- verbose
logical, if TRUE, the progress is shown
- ...
other arguments passed to
plot
Value
A matrix of expected heterozygote proportions from the observed data with p-value indicating significance of deviation.
Examples
if (FALSE) data(alleleINF)
AI <- alleleINF
duplicates<-sig.hets(AI,plot=TRUE,Fis=0.1) # \dontrun{}
#> assessing excess of heterozygotes